Publications
Full list below · Google Scholar ↗
Highlights

Revisiting the Nature of Low-Lying π Resonances in Micro-solvated Uracil
Low-energy electron interactions with biomolecules play a central role in radiation-induced chemistry. Initially, electrons are attached to the building blocks of biomolecules, leading to electronic resonances. Here, we present a theoretical investigation of π resonances in microsolvated uracil anion clusters using a combination of multireference and equation-of-motion coupled-cluster electronic structure methods. In addition to the bound anionic ground state, we identify three resonances. Our results are in excellent agreement with recent experiments, predicting a progressive stabilization of all anionic states with increasing numbers of water molecules, while the excitation energies of the anion remain almost constant with solvation. Importantly, our calculations show that the second low-lying π resonance has two-particle one-hole Feshbach resonance character, rather than the one-particle shape character assigned in prior experimental interpretation. Distinguishing between shape and Feshbach resonances is crucial for understanding low-energy electron–molecule interactions, as these states exhibit very different electronic structures, lifetimes, and, therefore, decay mechanisms. Overall, these findings highlight the essential role of high-level electronic-structure theory in interpreting transient states of anions in microsolvated biomolecular systems.
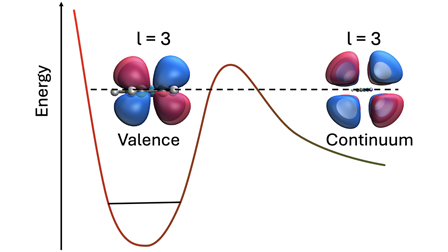
Partial Widths of Shape Resonances in Pyridine and Uracil Using the Stabilization Method
Low-energy electron attachment to molecules often leads to the formation of shape resonances, which play a pivotal role in electron-driven chemical processes. While the total decay width of a resonance determines its auto-detachment lifetime, decomposing this width into partial contributions from various auto-detachment continuum channels may provide a deeper insight into the underlying decay dynamics. In this work, we explore the applicability of using bound state methods, in particular the analytic-continuation based stabilization method, for determining partial widths in medium-sized organic molecules. Angular momentum-resolved partial widths can be obtained by placing diffuse functions at the molecular center of mass. Using the stabilization method combined with the equation-of-motion electron attachment coupled cluster method, we applied this technique to pyridine and uracil, two prototypical π-conjugated systems, and analyzed the contributions of s-, p-, d-, f-, g-, h-, and i-type functions to the widths of shape resonances. Our results show that the dominant angular momentum component of each resonance width correlates strongly with the nodal structure of the corresponding resonant orbital. Importantly, we find that higher angular momentum functions, particularly d, f, g, and h, play a decisive role in accurately capturing resonance widths. Compared to conventional atom-centered augmentation schemes, the center of mass-based approach alleviates some of the uncertainties in the stabilization method associated with inconsistent avoided crossings.

Impact of solvation on the electronic resonances in uracil
In this work, we study electron attachment to solvated uracil, an RNA nucleobase, using the orbital stabilization method at the Equation of Motion-Coupled Cluster for Electron Affinities with Singles and Doubles (EOM-EA-CCSD) level of theory with the Effective Fragment Potential (EFP) solvation method. We benchmarked the approach using multireference methods, as well as by comparing EFP and full quantum calculations. The impact of solvation on the first one particle (1p) shape resonance, formed by electron attachment to the π* LUMO orbital, as well as the first two particle one hole (2p1h) resonance, formed by electron attachment to neutral uracil's π–π* excited state, was investigated. We used molecular dynamics simulations for solvent configurations and applied charge stabilization technique-based biased sampling to procure configurations adequate to cover the entire range of the electron attachment energy distribution. The solvent effects were similar for the two resonances, indicating that the exact electron density of the state is not as important as the solvent configurations. Multireference calculations extended the findings showing that solvation effects are similar for the lowest four resonances, further indicating that the specific solute electron density is not as important, but rather the water configurations play the most important role in solvation effects. Finally, by comparing bulk solvation to clusters of uracil with a few water molecules around it, we find that the impact of microsolvation is very different from that of bulk solvation.
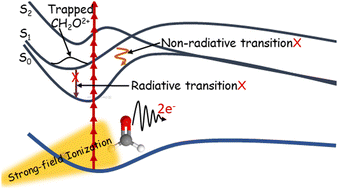
Stable excited dication: trapping on the S1 state of formaldehyde dication after strong field ionization
Combined theoretical and experimental work examines the dynamics of dication formaldehyde produced by strong field ionization. Trajectory surface hopping dynamics on the first several singlet electronic states of the formaldehyde dication are used to examine the relaxation pathways and dissociation channels, while kinetic energy distributions after strong field ionization of formaldehyde and deuterated formaldehyde are used to confirm the theoretical predictions. We find that the first excited state of the formaldehyde dication is stable, neither decays to the ground state nor dissociates, even though the ground state and higher lying states are directly dissociative. The stability of the first excited state is explained by its symmetry which does not allow for radiative or nonradiative transitions to the ground state and by large barriers to dissociate on the excited state surface.
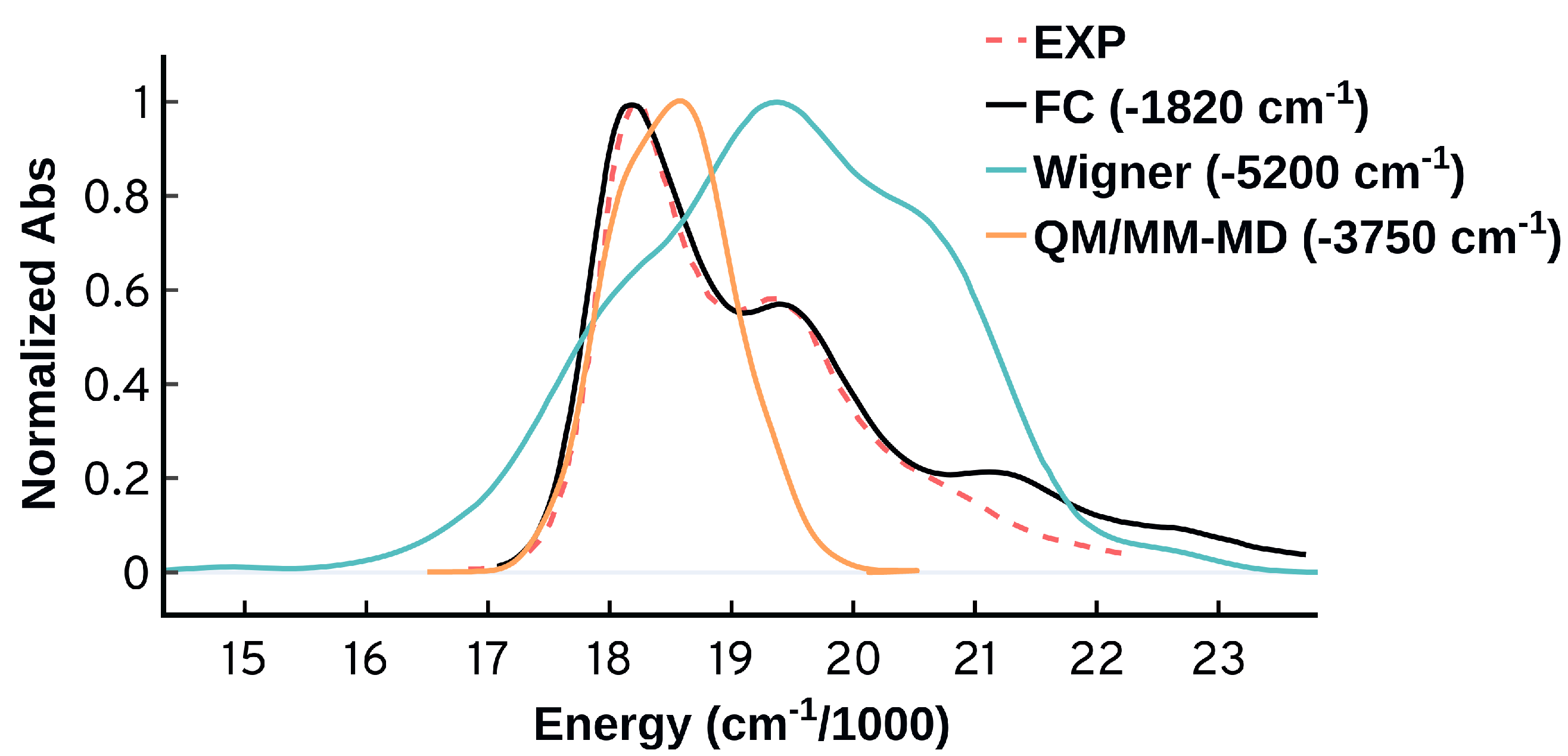
Modeling the Electronic Absorption Spectra of the Indocarbocyanine Cy3
Accurate modeling of optical spectra requires careful treatment of the molecular structures and vibronic, environmental, and thermal contributions. The accuracy of the computational methods used to simulate absorption spectra is limited by their ability to account for all the factors that affect the spectral shapes and energetics. The ensemble-based approaches are widely used to model the absorption spectra of molecules in the condensed-phase, and their performance is system dependent. The Franck–Condon approach is suitable for simulating high resolution spectra of rigid systems, and its accuracy is limited mainly by the harmonic approximation. In this work, the absorption spectrum of the widely used cyanine Cy3 is simulated using the ensemble approach via classical and quantum sampling, as well as, the Franck–Condon approach. The factors limiting the ensemble approaches, including the sampling and force field effects, are tested, while the vertical and adiabatic harmonic approximations of the Franck–Condon approach are also systematically examined. Our results show that all the vertical methods, including the ensemble approach, are not suitable to model the absorption spectrum of Cy3, and recommend the adiabatic methods as suitable approaches for the modeling of spectra with strong vibronic contributions. We find that the thermal effects, the low frequency modes, and the simultaneous vibrational excitations have prominent contributions to the Cy3 spectrum. The inclusion of the solvent stabilizes the energetics significantly, while its negligible effect on the spectral shapes aligns well with the experimental observations.
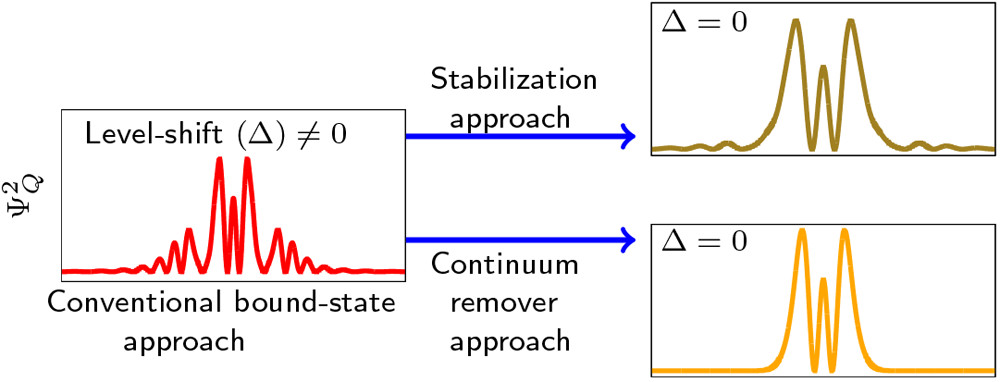
A unique QP-partitioning and Siegert width using real-valued continuum remover potential
A simple, practical quantum chemical procedure is presented for computing the energy position and the decay width of autoionization resonances. It combines the L2-stabilized resonance wave function obtained using the real-valued continuum-remover (CR) potential [Y. Sajeev Chem. Phys. Lett. 2013, 587, 105–112] and the Feshbach projection operator (FPO) partitioning technique. Unlike the conventional FPO partitioning of the total wave function into its resonant Q space and background P space components, an explicit partitioning of the total wave function into its interaction region and noninteraction region components is obtained with the help of real-valued continuum-remover potential. The molecular system is initially confined inside a CR potential which removes the electronic continuum of the molecular system in which its resonance state is embedded and, thus, unravels the Q space component of the resonance wave function as a bound, localized eigenstate of the confined system. The eigenfunctions of the molecular Hamiltonian represented in the {1-Q} space constitute a complementary, P orthogonal space. A unique QP partition is obtained when the level-shift of the Q space function due to its coupling with the P space is zero, and the resonance width is computed using these unique partitioned spaces.
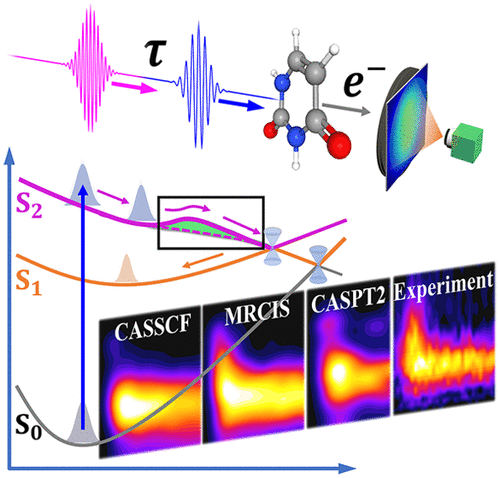
Time Resolved Photoelectron Spectroscopy as a Test of Electronic Structure and Nonadiabatic Dynamics
We compare different levels of theory for simulating excited state molecular dynamics and use time-resolved photoelectron spectroscopy measurements to benchmark the theory. We perform trajectory surface hopping simulations for uracil excited to the first bright state (ππ*) using three different levels of theory (CASSCF, MRCIS, and XMS-CASPT2) in order to understand the role of dynamical correlation in determining the excited state dynamics, with a focus on the coupling between different electronic states and internal conversion back to the ground state. These dynamics calculations are used to simulate the time-resolved photoelectron spectra. The comparison of the calculated and measured spectra allows us to draw conclusions regarding the relative insights and quantitative accuracy of the calculations at the three different levels of theory, demonstrating that detailed quantitative comparisons of time-resolved photoelectron spectra can be used to benchmark methodology.
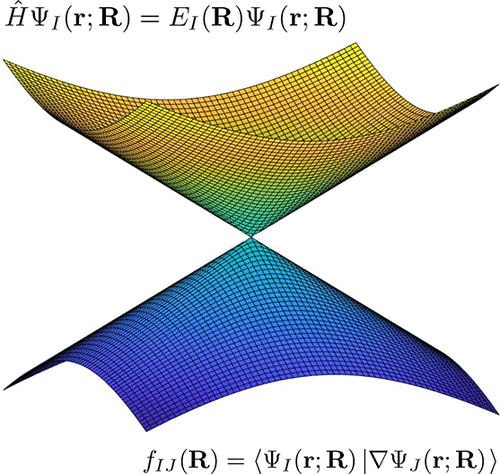
Electronic structure methods for the description of nonadiabatic effects and conical intersections
Nonadiabatic effects are ubiquitous in photophysics and photochemistry, and therefore, many theoretical developments have been made to properly describe them. Conical intersections are central in nonadiabatic processes, as they promote efficient and ultrafast nonadiabatic transitions between electronic states. A proper theoretical description requires developments in electronic structure and specifically in methods that describe conical intersections between states and nonadiabatic coupling terms. This review focuses on the electronic structure aspects of nonadiabatic processes. We discuss the requirements of electronic structure methods to describe conical intersections and nonadiabatic couplings, how the most common excited state methods perform in describing these effects, and what the recent developments are in expanding the methodology and implementing nonadiabatic couplings.
Understanding the Interplay Between the Non-Valence and Valence State of the Uracil Anion Upon Mono-Hydration
In this work we present an ab initio investigation into the effect of monohydration on the interaction of uracil with low energy electrons. Electron attachment and photodetachment experimental studies have previously shown dramatic changes in uracil upon solvation with even a single water molecule, due to an inversion of the character of the ground state of the anion. Here we explore the interplay between the nonvalence and valence states of the uracil anion, as a function of geometry and site of solvation. Our model provides unambiguous interpretation of previous photoelectron studies, reproducing the binding energies and photoelectron images for bare uracil and a single isomer of the U•(H2O)1 cluster. The results of this study provide insight into how electrons may attach to hydrated nucleobases. These results lay the foundations for further investigations into the effect of microhydration on the electronic structure and electron capture dynamics of nucleobases.
Full List
Exploring active learning strategies for excited state dynamics: Application to Uracil
Revisiting the Nature of Low-Lying π Resonances in Micro-solvated Uracil
Time resolved probing of the ultrafast excited state dynamics of cis, cis-1, 3-cyclooctadiene
Partial Widths of Shape Resonances in Pyridine and Uracil Using the Stabilization Method
Statistical vs. direct dissociation of molecular dications
How excitation wavelength affects excited state dynamics in o-nitrophenol: A theoretical perspective
COLUMBUS — an Efficient and General Program Package for Ground and Excited State Computations Including Spin-Orbit Couplings and Dynamics
Impact of solvation on the electronic resonances in uracil
Ultrafast structural dynamics of UV photoexcited cis,cis-1,3- cyclooctadiene observed with femtosecond electron diffraction
Modeling the Effect of Substituents on the Electronically Excited States of Indole Derivatives
Detecting Centrosymmetric Molecular Ions at an Interface with Vibrational Sum Frequency Generation Spectroscopy
Exploring electronic resonances in pyridine: Insights from orbital stabilization techniques
Time-resolved photoelectron spectroscopy via trajectory surface hopping
Spectroscopic approaches for studies of site-specific DNA base and backbone breathing using exciton-coupled dimer-labeled DNA
Nucleic Acids and Molecular Biology”, vol. 36, page 157-209, Springer, 2024
Molecular Dynamical and Quantum Mechanical Exploration of the Site-Specific Dynamics of Cy3 dimers internally linked to dsDNA
Quantum contributions to Coulomb-explosion imaging revealed by trajectory-surface-hopping molecular dynamics
Excited State Hydrogen or Proton Transfer Pathways in microsolvated n-cyanoindole fluorescent probes
Using transition density models to interpret experimental optical spectra of exciton-coupled cyanine (iCy3)2 dimer probes of local DNA conformations at or near functional protein binding sites
Unravelling the Origin of the Vibronic Spectral Signatures in an Excitonically Coupled Indocarbocyanine Cy3 Dimer
Excited State Dynamics of o-Nitrophenol Studied with UV Pump VUV Probe Time Resolved Photoelectron and Photoion Spectroscopy
Mechanistic aspects of the effect of flanking nucleotide sequence on CPD formation and CPD self-repair in DNA
Spectroscopy and Theoretical Modeling of Tetracene Anion Resonances
Effective Fragment Potentials for Microsolvated Excited and Anionic States
Strong Field Double Ionization of Formaldehyde Investigated using Momentum Resolved Covariance Imaging and Trajectory Surface Hopping
Nonadiabatic Excited State Dynamics of Organic Chromophores: Take-Home Messages
Stable excited dication: trapping on the S1 state of formaldehyde dication after strong field ionization
Modeling the Electronic Absorption Spectra of the Indocarbocyanine Cy3
Conformer-Specific Dissociation Dynamics in Dimethyl Methylphosphonate Radical Cation
Projected Complex Absorbing Potential Multi-Reference Configuration Interaction for Shape and Feshbach Resonances
A unique QP-partitioning and Siegert width using real-valued continuum remover potential
Accurate Modeling of Excitonic Coupling in Cyanine Dye Cy3
Modeling the Ultrafast Electron Attachment Dynamics of Solvated Uracil
Time Resolved Photoelectron Spectroscopy as a Test of Electronic Structure and Nonadiabatic Dynamics
Electronic structure methods for the description of nonadiabatic effects and conical intersections
Modeling Solvation Effects on Absorption and Fluorescence Spectra of Indole in Aqueous Solution
Benchmarking Quantum Mechanical Methods for the Description of Charge-Transfer States in π Stacked Nucleobases
Effect of Dynamic Correlation on the Ultrafast Relaxation of Uracil in the Gas Phase
Description of Two-particle One-hole Electronic Resonances using Orbital Stabilization Methods
Understanding the Interplay Between the Non-Valence and Valence State of the Uracil Anion Upon Mono-Hydration
Stabilization of triplet biradical intermediate of 5-methylcytosine enhances cyclobutane pyrimidine dimer (CPD) formation in DNA
Excited State Dynamics of cis,cis-1,3-Cyclooctadiene: UV Pump VUV Probe Time Resolved Photoelectron Spectroscopy
Excited State Dynamics of cis,cis-1,3-Cyclooctadiene: Non-adiabatic Trajectory Surface Hopping
The Generality of the GUGA MRCI Approach in COLUMBUS for Treating Complex Quantum Chemistry
Comparative study of methodologies for calculating metastable states of small to medium-sized molecules
Electron correlation in channel resolved strong field molecular double ionization
Theoretical Investigation of Positional Substitution and Solvent Effects on n-Cyanoindole Fluorescent Probes
Role of charge transfer states into the formation of cyclobutane pyrimidine dimers in DNA
Intersystem crossing in the exit channel
Strong and Weak-Field Ionization in Pump-Probe Spectroscopy
Ultrafast Photoinduced Processes in Polyatomic Molecules: Electronic Structure, Dynamics and Spectroscopy dedicated to Prof. Wolfgang Domcke on the occasion of his 70th birthday
Introduction: Theoretical Modeling of Excited State Processes
The Origin of Fluorescence in DNA Thio-Analogues
Electron-induced origins of prebiotic sugars: self-reactions of methanol anion clusters
Electronic Resonances of Nucleobases Using Stabilization Methods
Quadruple coincidence measurement of electron correlation in strong field molecular double ionization
Photochemical Formation of Cyclobutane Pyrimidine Dimers in DNA through Electron Transfer from a Flanking Base
Mechanistic insights into photoinduced damage of DNA and RNA nucleobases in the gas phase and in bulk solution
Origins of Photodamage in Pheomelanin Constituents: Photochemistry of 4-Hydroxybenzothiazole
Calculations of non-adiabatic couplings within equation-of-motion coupled-cluster framework: Theory, implementation, and validation against multi-reference methods
Vibrationally Assisted Below Threshold Ionization
Mechanisms of H and CO Loss from the Uracil Anion Following Low Energy Electron Irradiation
Controlling Photorelaxation in Uracil with Shaped Laser Pulses: A Theoretical Assessment
Ultrafast Internal Conversion Dynamics of Highly Excited Pyrrole Studied with VUV/UV Pump Probe Spectroscopy
Substituent Effects on the Absorption and Fluorescence Properties of Anthracene
Conformational and electronic effects on the formation of anti cyclobutane pyrimidine dimer in G-quadruplex structure
Core-Excited and Shape Resonance of Uracil
Coexistence of different electron transfer mechanisms in the DNA repair process by photolyase
Photophysical Properties of Pyrrolocytosine, a Cytosine Fluorescent Base Analogue
Molecular Double Ionization Using Field Few Cycle Laser Pulses
Excimers and Exciplexes in Photoinitiated Processes of Oligonucleotides
J. Phys. Chem. Lett., invited perspective, 7, 976-984, (2016)
Surface hopping investigation of the relaxation dynamics in radical cations
Photophysical Deactivation Pathways in Adenine Oligonucleotides
Controlling the Dissociation Dynamics of Acetophenone Radical Cation Through Excitation of Ground and Excited State Wavepackets
Journal of Physics B Atomic, Molecular and Optical Physics, 48, 164002, (2015)
Excited state relaxation of neutral and basic 8-Oxoguanine
QM/MM studies reveal pathways leading to the quenching of the formation of thymine dimer photoproduct by flanking bases
Photoelectron spectrum and dynamics of the uracil cation
Strong Field Adiabatic Ionization Prepares a Launch State for Coherent Control
Tribute to David R. Yarkony
What We Can Learn from the Norms of One-particle Density Matrices, and What We Can't
Role of Excitonic Coupling and Charge-Transfer States in the Absorption and CD Spectra of Adenine-Based Oligonucleotides Investigated through QM/MM Simulations
Theoretical Studies of the Excited States of p-Cyanophenylalanine and Comparisons with the Natural Amino Acids Phenylalanine and Tyrosine
Radical Cation Spectroscopy of Substituted Alkyl Phenyl Ketones via Tunnel Ionization
Ultrafast Excited-State Dynamics and Vibrational Cooling of 8-oxo-7,8-dihydro-2-deoxyguanosine in D2O
Measurement of Ionic Resonances in Alkyl Phenyl Ketone Cations via Infrared Strong Field Mass Spectrometry
Ultrafast Relaxation Dynamics of Uracil Probed via Strong Field Dissociative Ionization
Excited-State Tautomerization of Gas-Phase Cytosine
Dissociative electron attachment to carbon dioxide via the 2Πu shape resonance
Measurement of an Electronic Resonance in Ground State, Gas Phase Acetophenone Cation via Strong Field Mass Spectrometry
High-Multiplicity Natural Orbitals in Multireference Configuration Interaction for Excited State Potential Energy Surfaces
Exciplexes and conical intersections lead to fluorescence quenching in pi-stacked dimers of 2-aminopurine with nucleobases
Contrasting Photophysical Behaviors of Star-shaped vs Linear Chromophores
A Benchmark of Excitonic Couplings Derived from Atomic Transition Charges
Angle-Resolved Strong Field Ionization of Polyatomic Molecules: More than the Orbitals Matters
Ultrafast Excited State Dynamics of Allopurinol, a Modified DNA Base
Neutral Ionic Correlations in Strong Field Molecular Ionization
Dyson Norms in XUV and Strong-field Ionization of Polyatomics: Cytosine and uracil
Fragmentation Pathways in the Uracil Radical Cation
Correction to Pathways for Fluorescence Quenching in 2-Aminopurine pi-Stacked with Pyrimidine Nucleobases
Two Dimensional Fourier-Transform Spectroscopy of Adenine and Uracil Using Shaped Ultrafast Laser Pulses in the Deep UV
On the Accessibility to Conical Intersections in Purines: Hypoxanthine and its Singly Protonated and Deprotonated Forms
High-multiplicity natural orbitals in multireference configuration interaction for excited states
Absorption, Circular Dichroism and Photoluminescence in Perylene Diimide Bichromophores: Polarization Dependent H- and J-aggregate Behavior
Nuclear Dynamics on a Three-state Jahn-Teller Model System
Following Ultrafast Radiationless Relaxation Dynamics With Strong Field Dissociative Ionization: A Comparison Between Adenine, Uracil, and Cytosine
IEEE Journal of selected topics in Quantum Electronics, 18, 187-194, (2012)
Strong Field Molecular Ionization from Multiple Orbitals
Combining dissociative ionization pump probe spectroscopy and ab initio calculations to explore excited state dynamics involving conical intersections
Distinguishing Between Relaxation Pathways by Combining Dissociative Ionization Pump Probe Spectroscopy and ab initio Calculations: A Case Study of Cytosine
Pathways for fluorescence quenching in 2-aminopurine pi-stacked with pyrimidine nucleobases
Nonadiabatic Events and Conical Intersections
Photophysical pathways of cytosine in aqueous solution
Excited State Energies and Electronic Couplings of DNA Base Dimers
Change in Electronic Structure upon Optical Excitation of 8-Vinyladenosine: An Experimental and Theoretical Study
An Ab Initio Study of Substituent Effects on the Excited States of Purine Derivatives
On the Electronically Excited States of Uracil
Three-State Conical Intersections in Cytosine and Pyrimidinone Bases
Interpreting Ultrafast Molecular Fragmentation Dynamics with ab initio Calculations
Two and three state conical intersections in uracil cation radical
2-Aminopurine Excited State Electronic Structure Measure by Stark Spectroscopy
Excited Electronic States and Photophysics of Uracil-Water Complexes
Inclusion of Second-order Correlation Effects for the Ground and Singly Excited States Suitable for the Study of Conical Intersections: the CIS(2) Model
Cytosine in Context: A Theoretical Study of Substituent Effects on the Excitation Energies of 2-Pyrimidinone Derivatives
6MAP, a fluorescent adenine analogue, is a probe of base flipping by DNA photolyase
Radiationless Decay Mechanism of Cytosine: An Ab Initio Study with Comparisons to the Fluorescent Analogue 5-Methyl-2-Pyrimidinone
The Fluorescence Mechanism of 5-Methyl-2-Pyrimidinone: An Ab Initio Study of a Fluorescent Pyrimidine Analog
Conical intersections in Molecular Systems
A Combined Multireference Configuration Interaction/Molecular Dynamics Approach for Calculating Solvatochromic Shifts- Application to the nO -> pi* Electronic Transition in Formaldehyde
Excited Electronic States of the Cyclic Isomers of O3 and SO2
Three-state Conical Intersections in Nucleic Acid Bases
Radiationless Decay of Excited States of Uracil through Conical Intersections
Quantitative Detection of Singlet O2 via Cavity Enhanced Absorption
Conical Intersections of Three Electronic States Affect the Ground State of Radical Species with Little or No Symmetry: Pyrazolyl
Beyond Two-State Conical Intersections. Three-State Conical Intersections in Low Symmetry Molecules: the Allyl Radical
Photodissociation of the Vinoxy Radical through Conical, and Avoided, Intersections
Accidental Conical Intersections of Three States of the Same Symmetry. I. Location and Relevance
Spin-Orbit Coupling and Conical Intersections . IV. A Perturbative Determination of the Electronic Energies, Derivative Couplings and a Rigorous Diabatic Representation near a Conical Intersection. The General Case
Conical Intersections and the Nonadiabatic Reactions H2O+O(3P) -> OH(A)+OH(X)
Conical Intersections and the Spin-Orbit Interaction
Intersecting Conical Intersection Seams: their Location, Representation, and Effect on Local Topography
Spin-Orbit Coupling and Conical Intersections in Molecules with an Odd Number of Electrons. III. A Perturbative Determination of the Electronic Energies, Derivative Couplings and a Rigorous Diabatic Representation near a Conical Intersection
On the Effects of Spin-Orbit Coupling on Conical Intersection Seams in Molecules with an Odd Number of Electrons. II. Characterizing the Local Topography of the Seam
On the Effects of Spin-Orbit Coupling on Conical Intersection Seams in Molecules with an Odd Number of Electrons. I. Locating the Seam
Electronic Structure and Spectra of Actinyl Ions
Actinyl Ions in Cs2UO2Cl4
Intensities in the Spectra of Actinyl Ions
The Electronic Spectrum of the NpO22+ and NpO2+ Ions
Atomic Orbital Basis Sets for Use with Effective Core Potentials
Spin-Orbit Splittings in Mg+-Neutral Complexes
Ab Initio Study of the Ground and Several Excited States of the NLi System