Research
Conical intersections and nonadiabatic effects
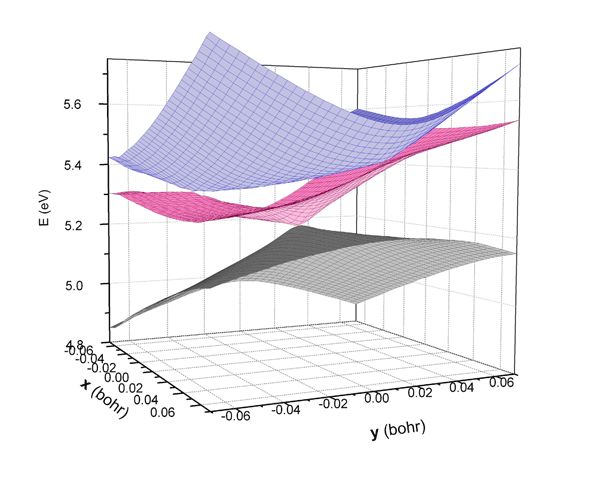 Most chemical processes follow the Born-Oppenheimer (adiabatic) approximation, in which the nuclei move on a single electronic potential energy surface (PES). However there are important processes where this approximation breaks down. These nonadiabatic events play an important role in essential processes in nature such as photosynthesis, vision, charge transfer and photochemistry. Nonadiabatic processes are facilitated by the close proximity of two or more PES where radiationless transitions between the surfaces can occur with higher probability. The efficiency for radiationless transitions increases in the extreme case when two PES become degenerate forming conical intersections. Through our work, as well as others, it has also been shown that three states can also become degenerate forming three-state conical intersections. Thus many ultrafast nonradiative transitions occur through conical intersections. Theoretical developments have enabled the efficient study of conical intersections in small systems. The focus of our group is to extend these studies to more complicated systems, particularly of biological interest, in an effort to understand the underlying mechanisms of photoinitiated nonadiabatic processes and their potential implications.
Most chemical processes follow the Born-Oppenheimer (adiabatic) approximation, in which the nuclei move on a single electronic potential energy surface (PES). However there are important processes where this approximation breaks down. These nonadiabatic events play an important role in essential processes in nature such as photosynthesis, vision, charge transfer and photochemistry. Nonadiabatic processes are facilitated by the close proximity of two or more PES where radiationless transitions between the surfaces can occur with higher probability. The efficiency for radiationless transitions increases in the extreme case when two PES become degenerate forming conical intersections. Through our work, as well as others, it has also been shown that three states can also become degenerate forming three-state conical intersections. Thus many ultrafast nonradiative transitions occur through conical intersections. Theoretical developments have enabled the efficient study of conical intersections in small systems. The focus of our group is to extend these studies to more complicated systems, particularly of biological interest, in an effort to understand the underlying mechanisms of photoinitiated nonadiabatic processes and their potential implications.
Photophysical behavior of DNA/RNA bases /Designing more efficient fluorescent probes
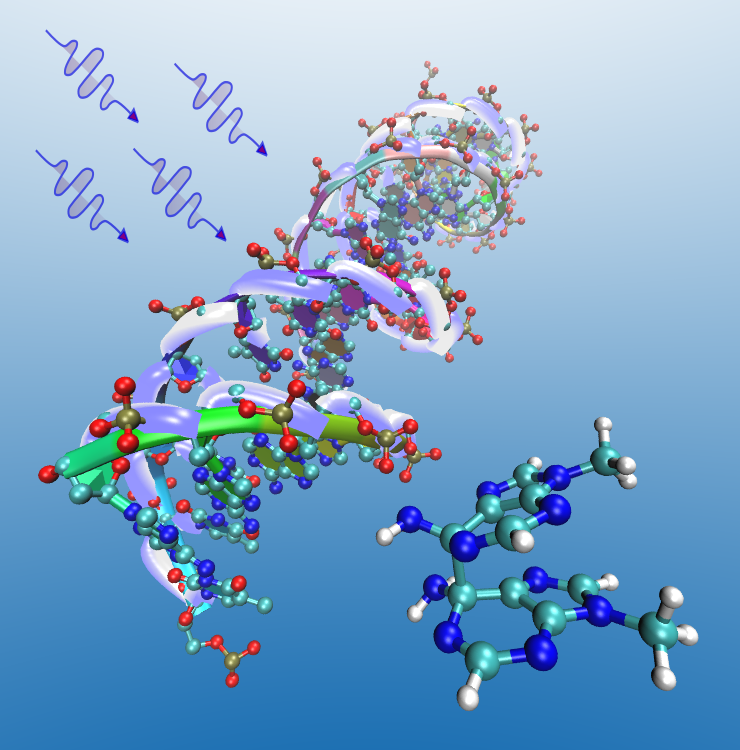 The photophysical and photochemical behavior of the nucleobases is of particular importance since they are the chromophores in DNA and RNA. The excited states of the nucleobases are extremely short-lived, having lifetimes of the order of femtoseconds or picoseconds. This property has been associated with their photostability in ultraviolet (UV) radiation from the sun. When UV radiation is absorbed by DNA photochemistry can occur which may lead to DNA damage and photocarcinogenesis. Efficient dissipation of the absorbed energy prevents extensive photodamage. We are investigating the radiationless decay mechanisms that can explain the ultrafast lifetimes and fluorescence quenching in these systems. Two- and three-state conical intersections are present that can facilitate nonadiabatic transitions between the different electronic states of these molecules.
Understanding the mechanism for fluorescence quenching can have important applications as well, since it enables us to design molecules that will not have this property, and they will instead fluoresce.
In recent work we have found exciting new insight into the excited state dynamics of pi-stacked nucleobases. These results may help us understand the more complex dynamics in single and double stranded DNA. Furthermore, we have been able to explain how quenching of the fluorescence occurs in some fluorescent probes when pi-stacked with other bases. These knowledge can help design more efficient fluorescent probes.
The photophysical and photochemical behavior of the nucleobases is of particular importance since they are the chromophores in DNA and RNA. The excited states of the nucleobases are extremely short-lived, having lifetimes of the order of femtoseconds or picoseconds. This property has been associated with their photostability in ultraviolet (UV) radiation from the sun. When UV radiation is absorbed by DNA photochemistry can occur which may lead to DNA damage and photocarcinogenesis. Efficient dissipation of the absorbed energy prevents extensive photodamage. We are investigating the radiationless decay mechanisms that can explain the ultrafast lifetimes and fluorescence quenching in these systems. Two- and three-state conical intersections are present that can facilitate nonadiabatic transitions between the different electronic states of these molecules.
Understanding the mechanism for fluorescence quenching can have important applications as well, since it enables us to design molecules that will not have this property, and they will instead fluoresce.
In recent work we have found exciting new insight into the excited state dynamics of pi-stacked nucleobases. These results may help us understand the more complex dynamics in single and double stranded DNA. Furthermore, we have been able to explain how quenching of the fluorescence occurs in some fluorescent probes when pi-stacked with other bases. These knowledge can help design more efficient fluorescent probes.
Understanding laser control of molecular dynamics
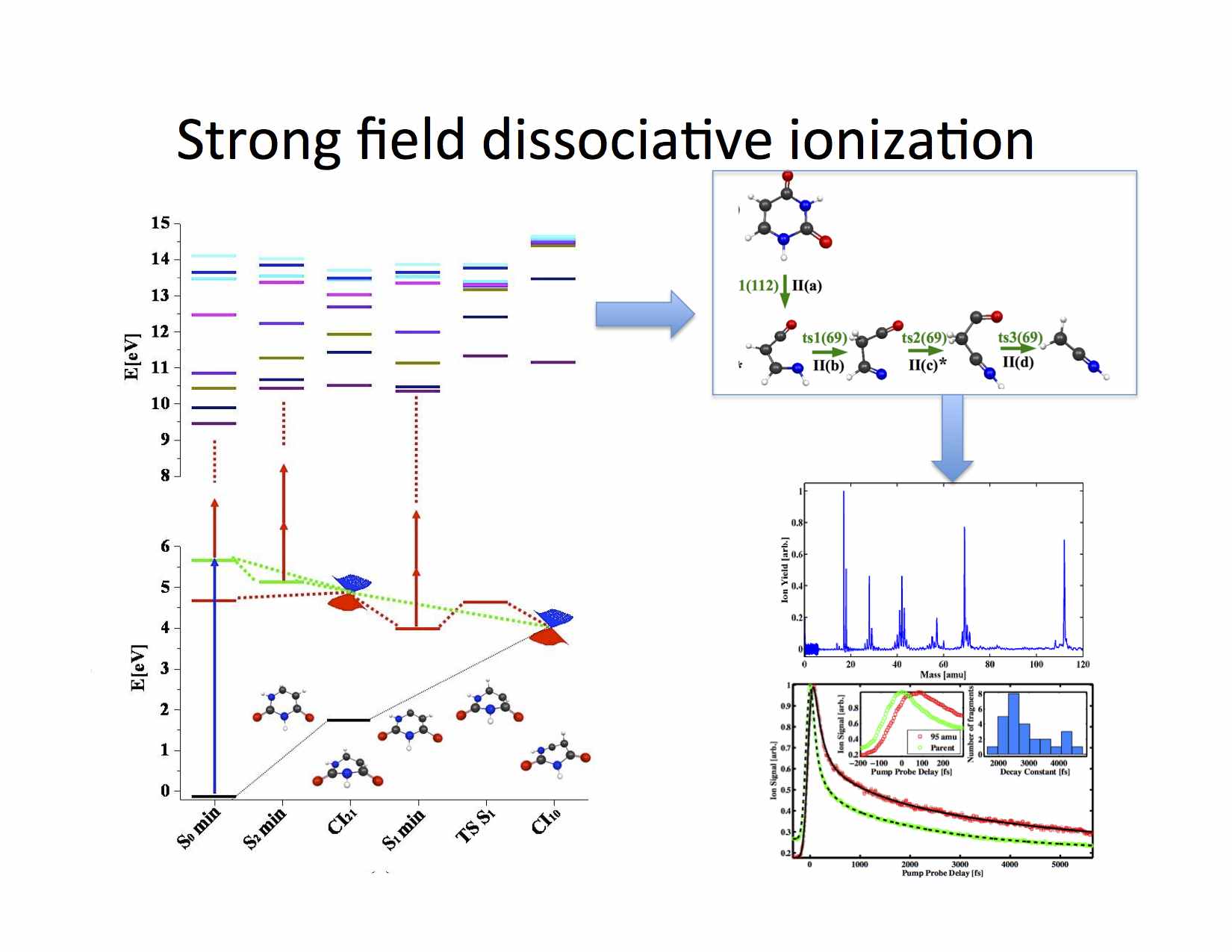 We are working in collaboration with Prof. Thomas Weinacht on a project aimed at understanding the underlying mechanisms involved in the dynamics and in laser control of basic chemical reactions, such as fragmentation and isomerization. Intense, shaped, ultrafast laser pulses are used to follow and control molecular dynamics and high level ab initio calculations to interpret the dynamics and guide the control. We are applying the techniques and understanding we have developed to dissociative ionization pump-probe spectroscopy and pulse shape spectroscopy. Close collaboration between theory and experiment is the major element in this project and this interaction has already lead to detailed understanding of several key processes. Theoretical calculations are used to examine the excited state dynamics of neutral systems, while calculations on the ion are used to find which fragmentation patterns after the probe which lead to the final signal. Key element is also the ionization process in strong laser fields, for which we have gained unique perspectives in recent work with other collaborators.
We are working in collaboration with Prof. Thomas Weinacht on a project aimed at understanding the underlying mechanisms involved in the dynamics and in laser control of basic chemical reactions, such as fragmentation and isomerization. Intense, shaped, ultrafast laser pulses are used to follow and control molecular dynamics and high level ab initio calculations to interpret the dynamics and guide the control. We are applying the techniques and understanding we have developed to dissociative ionization pump-probe spectroscopy and pulse shape spectroscopy. Close collaboration between theory and experiment is the major element in this project and this interaction has already lead to detailed understanding of several key processes. Theoretical calculations are used to examine the excited state dynamics of neutral systems, while calculations on the ion are used to find which fragmentation patterns after the probe which lead to the final signal. Key element is also the ionization process in strong laser fields, for which we have gained unique perspectives in recent work with other collaborators.
Development of methods and applications for electron-induced reactions
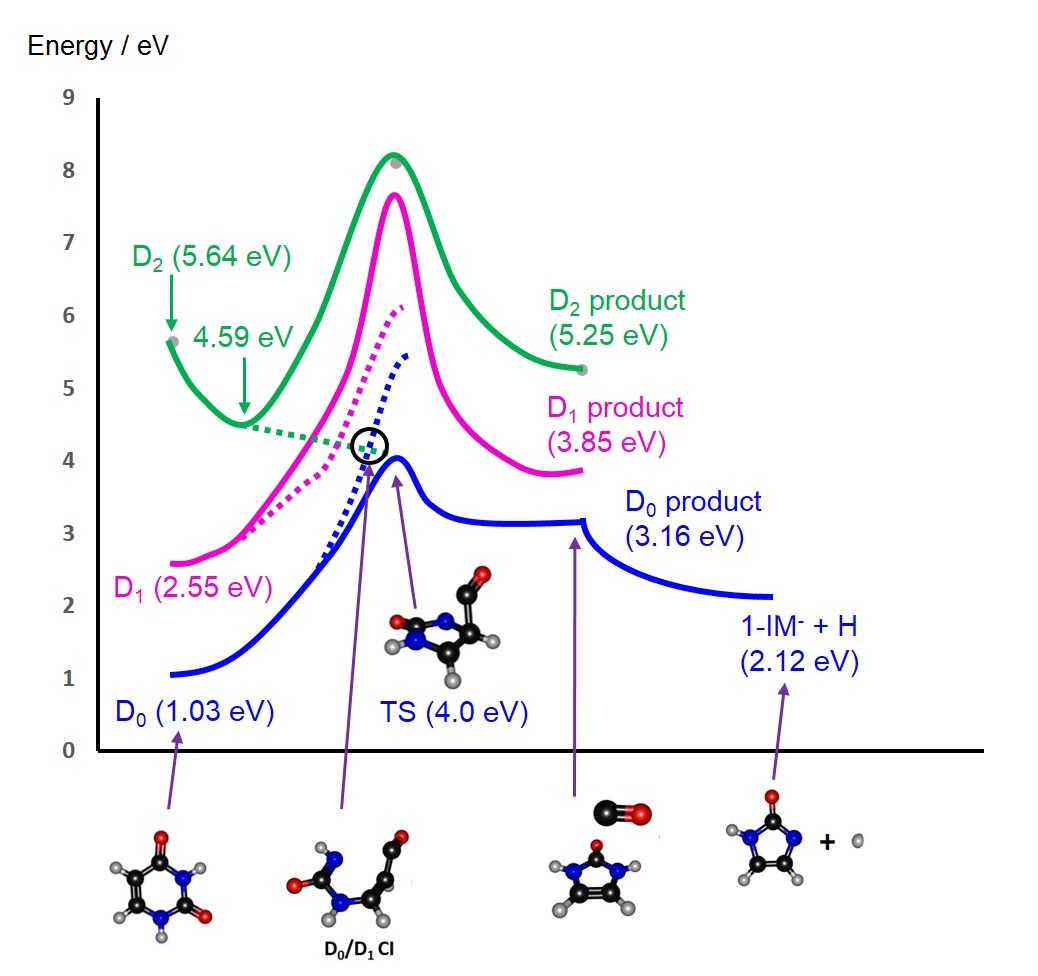 Electron-induced reactions are important in a myriad of chemical and biological process (including human physiology, cosmology and medicine). Electron attachment to a closed-shell neutral molecule usually represents a net thermodynamic destabilization - leading to a metastable anion resonance that is heavily coupled to the continuum. States within the continuum are unstable with respect to electron detachment – thereby driving the system towards the parent ground state neutral + electron at an infinite separation. Despite this finite electron detachment lifetime, experiments have observed a diverse range of electron-induced reactions following irradiation with electrons with well-defined kinetic energies. The clear competition between such electron-induced reactions and electron detachment is of particular interest to our group but is computational challenging. For example, the resonance/continuum coupling means that the associated electronic wavefunction is continuous. The continuous wavefunction is therefore non-square integrable and thus cannot be computed with mainstream electronic structure theory. Our group is therefore interested in modifying existing electronic structure methods and developing complex Hamiltonian methods that ‘stabilize’ a given anion resonance position. Using such developments, we are particularly interested in the nuclear motions that drive electron-induced reactions. Such methods were recently used to map the potential energy profiles associated with the dissociative electron attachment of Uracil observed experimentally (see figure). The interaction of low energy electrons with nucleobases is of particular importance because it can lead to biological damage.
Electron-induced reactions are important in a myriad of chemical and biological process (including human physiology, cosmology and medicine). Electron attachment to a closed-shell neutral molecule usually represents a net thermodynamic destabilization - leading to a metastable anion resonance that is heavily coupled to the continuum. States within the continuum are unstable with respect to electron detachment – thereby driving the system towards the parent ground state neutral + electron at an infinite separation. Despite this finite electron detachment lifetime, experiments have observed a diverse range of electron-induced reactions following irradiation with electrons with well-defined kinetic energies. The clear competition between such electron-induced reactions and electron detachment is of particular interest to our group but is computational challenging. For example, the resonance/continuum coupling means that the associated electronic wavefunction is continuous. The continuous wavefunction is therefore non-square integrable and thus cannot be computed with mainstream electronic structure theory. Our group is therefore interested in modifying existing electronic structure methods and developing complex Hamiltonian methods that ‘stabilize’ a given anion resonance position. Using such developments, we are particularly interested in the nuclear motions that drive electron-induced reactions. Such methods were recently used to map the potential energy profiles associated with the dissociative electron attachment of Uracil observed experimentally (see figure). The interaction of low energy electrons with nucleobases is of particular importance because it can lead to biological damage.